PiHKAL: The Chemical Story 9

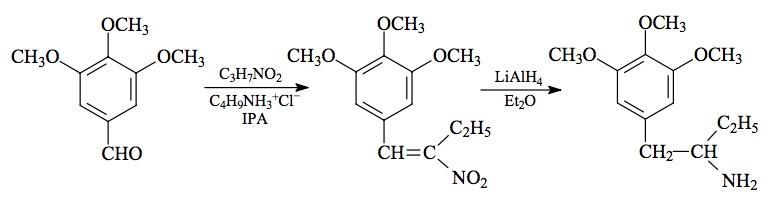
#106 MDE; MDEA; EVE; N-ETHYL-MDA; 3,4-METHYLENEDIOXY-N-ETHYLAMPHETAMINE

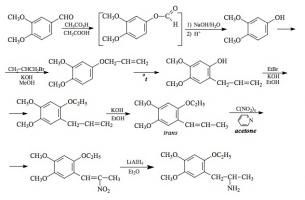
SYNTHESIS: (from MDA) To a solution of 3.6 g of the free base of 3,4-methylenedioxyamphetamine (MDA) in 20 g pyridine, there was added 2.3 g acetic anhydride, and the mixture stirred at room temperature for 0.5 h. This was then poured into 250 mL H2O and acidified with HCl. This aqueous phase was extracted with 3x75 mL CH2Cl2, the extracts pooled and washed with dilute HCl, and the solvent removed under vacuum. The pale amber residue of N-acetyl-3,4-methylenedioxyamphetamine weighed 5.2 g as the crude product, and it was reduced without purification. On standing it slowly formed crystals. Recrystallization from a mixture of EtOAc/hexane (1:1) gave white crystals with a mp of 92-93 °C.
A stirred suspension of 4.8 g LAH in 400 mL anhydrous THF was brought up to a reflux, and then treated with a solution of 5.0 g of the impure N-acetyl-3,4-methylenedioxyamphetamine in 20 mL anhydrous THF. Reflux conditions were maintained for 3 days, and then after cooling in an ice bath, the excess hydride was destroyed with the careful addition of H2O. The 4.8 mL H2O (in a little THF) was followed with 4.8 mL of 15% NaOH, and finally an additional 15 mL H2O. The white, granular, basic mass of inorganic salts was removed by filtration, the filter cake washed with additional THF, and the combined filtrate and washings stripped of solvent under vacuum. The residue was dissolved in 20 mL IPA, made acidic with 40 drops of concentrated HCl, and diluted with 150 mL anhydrous Et2O. The crystalline product was removed by filtration, washed with 80% Et2O (containing IPA) followed by Et2O itself, and then air dried to provide 3.0 g of 3,4-methylenedioxy-N-ethylamphetamine hydrochloride (MDE) as fine white crystals with a mp of 198-199 °C.
(from 3,4-methylenedioxyphenylacetone with aluminum amalgam) To 40 g of thin aluminum foil cut in 1 inch squares (in a 2 L wide mouth Erlenmeyer flask) there was added 1400 mL H2O containing 1 g mercuric chloride. Amalgamation was allowed to proceed until there was the evolution of fine bubbles, the formation of a light grey precipitate, and the appearance of occasional silvery spots on the surface of the aluminum. This takes between 15 and 30 min depending on the freshness of the surfaces and the temperature of the H2O. The H2O was removed by decantation, and the aluminum was washed with 2x1400 mL of fresh H2O. The residual H2O was removed as thoroughly as possible by shaking, and there was added, in succession and with swirling, 72.5 g ethylamine hydrochloride dissolved in 60 mL warm H2O, 180 mL IPA, 145 mL 25% NaOH, 53 g 3,4-methylenedioxy-phenylacetone (see under MDMA for its preparation), and finally 350 mL IPA. The exothermic reaction was kept below 60 °C with occasional immersion into cold water and, when it was thermally stable, it was allowed to stand until it had returned to room temperature and all the insolubles settled to the bottom as a grey sludge. The clear yellow overhead was decanted and the sludge removed by filtration and washed with MeOH. The combined decantation, mother liquors, and washes, were stripped of solvent under vacuum, the residue suspended in 1500 ml of H2O, and sufficient HCl added to make the phase distinctly acidic. This was then washed with 2x100 mL CH2Cl2, made basic with 25% NaOH, and extracted with 3x100 mL of CH2Cl2. After removal of the solvent from the combined extracts, there remained 59.5 g of an amber oil which was distilled at 145-150 °C at 0.5 mm/Hg, producing 40.3 g of an off-white oil. This was dissolved in 600 mL IPA, neutralized with about 20 mL of concentrated HCl and then treated with 300 mL anhydrous Et2O. After filtering off the white crystals, washing with a IPA/Et2O (2:1) mixture, with Et2O and air drying, the final 3,4-methylenedioxy-N-ethylamphetamine hydrochloride (MDE) weighed 37.4 g.
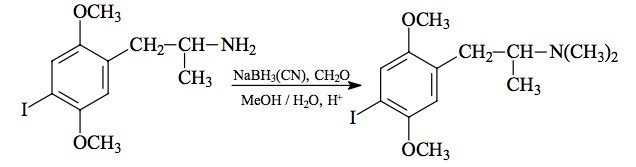
(from 3,4-methylenedioxyphenylacetone with NaBH3CN) To a well stirred solution of 31.0 g ethylamine hydrochloride in 110 mL MeOH there was added 6.6 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 3.0 g sodium cyanoborohydride. Concentrated HCl in MeOH was added as required to maintain the pH at about 6 as determined with external, dampened universal pH paper. About 2 days were required for the reduction to be complete as determined by the final stabilization of the pH. The reaction mixture was added to 1 L H2O and made strongly acidic with an excess of HCl. After washing with 2x100 mL CH2Cl2 the aqueous phase was made basic with 25% NaOH, and extracted with 3x100 mL CH2Cl2. Removal of the solvent under vacuum yielded 8.3 g of a pale amber oil that was distilled at 85-100 °C at 0.2 mm/Hg. There was obtained 6.0 g of a water-white oil that was dissolved in 65 mL IPA and neutralized with 75 drops of concentrated HCl which produced crystals spontaneously. These were diluted with some 20 mL of anhydrous Et2O removed by filtration, washed first with IPA/Et2O (2:1), and then with Et2O. After air drying there was obtained 6.1 g of 3,4-methylenedioxy-N-ethylamphetamine hydrochloride (MDE) with a mp of 201-202 °C. Anal. (C12H18ClNO2) N.
DOSAGE: 100 - 200 mg.
DURATION: 3 - 5 h.
QUALITATIVE COMMENTS: (with 100 mg) There was a warm light all about me. And a gentle, almost alcohol-like, intoxication. The drug seems to change my state of awareness, but it does nothing else. The world is as intense or as dull as I choose to make it. At the 1.5 hour point I was clearly dropping, and an hour later yet, completely without residue.
(with 160 mg) The first effects were felt in forty minutes and I seemed to be completely there by the end of that first hour. There was an initial slightly dizzy intoxication, and then I felt very nice. A good intoxication, with maybe a little motor incoordination. There was absolutely no appetite at all. The next morning there was still some feeling of elation but I was still very relaxed. High marks for the quality of the experience.
(with 160 mg) Overall this was a wonderful experience. I felt that the effect was stronger and smoother than MDMA, but perhaps the group enhancement may be partly responsible. I felt definitely fewer physiological side-effects than with MDMA, particularly the urinating problem; although there was dehydration, there was less burning annoyance.
(with 160 mg) I was hard hit, to the extent that there was difficulty in verbalizing and following other people's thoughts. I entered the experience with some cold symptoms, and my sore throat disappeared. I felt quite intoxicated and tranquilized.
(with 200 mg) Very stoned. There was some nausea in the beginning of the experience. As it developed I found it very difficult to concentrate on what I was thinking or saying simply due to the extraordinary nature of coming on to this material. There is noticeable jaw-clenching and rice crispies in the ears. This is a meditative material not unlike MDMA except there are more difficulties in forming words. And there is a problem in focusing the eyes, what I want to call Teye-romp.' My anorexia was extremely long-lived Q perhaps a total of 72 hours. This may have been too high a dosage.
EXTENSIONS AND COMMENTARY: This immediate homologue of MDMA has a very similar chronology but requires a slightly larger dose. Another similarity is the occasional report of teeth clenching, especially following the use of supplemental dosages intended to extend the effects of the drug. These supplements have been explored in the 50 to 75 milligram range, usually at the two hour point. In one unpublished clinical experiment with MDMA, an extension was attempted at the 1 hour 45 minute point with MDE rather than with MDMA, to see if there was any change in the qualitative character of the experience. The effective time of intoxication was extended, but the group fell surprisingly quiet, with a drop in the usual urge to converse and interact.
The effects of MDE are similar in many ways to those of MDMA, but there are believable differences. The particular magic, and affective transference, does not appear to be there. There is a stoning intoxication, as there is with MDA, and there is a seemingly unrewarding aspect to the upping of the dosages, again similar to MDA, and the properties of unusually easy communication and positive self-viewing of MDMA seem to be absent. Maybe the RSS isomer would have these properties, and they are lost in the racemate due to something coming from a more potent "intoxicating" RRS isomer. The optical isomers have never been evaluated separately in man.
There are only two ways in which two drugs can interact to produce a result that is not obvious from the summing of their individual actions. One is the process of synergism, where two active materials are allowed to interact within a single individual and at one time, and the consequence of this interaction is different than that which would have been expected. The other is the process of potentiation, where only one drug is active, but the presence of the second (and inactive) drug enhances the observed action of the first. MDE seems to fall in the first category.
The "piggy-back" or "window exploitation" studes were first discovered and explored with MDE, and have subsequently been extended most successfully with MDMA. The earliest procedure used was to assay modest quantities of active materials at the drop-off period of MDE, to exploit the open and benign state that was present. Usually, only a fraction of the standard dosage of the following drug was necessary to evoke a full experience. In psychotherapy applications, this sequence has been frequently used with MDMA followed by a second material that has been chosen to modify and expand the opening that the MDMA produced.
With the placement of MDMA under legal control in 1985, MDE occasionally appeared in the illicit street trade. It had been called EVE, which carries some perverse logic in light of the nickname used occasionally for MDMA, which was ADAM. The term INTELLECT has been used for it as well, but there has been no apparent reason advanced for this. And a final note on nomenclature. An old literature use of the code MDE was for the compound 3,4-methylenedioxyethanol-amine. See the discussion on this under the recipe for DME.
I have been told of an analogue of MDE that has been synthesized, and explored by the researcher who synthesized it. It contains the N-trifluoroethyl group common to several pharmaceuticals such as Quazepam. The analogue is 3,4-methylenedioxy-N-(2,2,2-trifluoroethyl)amphetamine hydrochloride (mp 207-209 °C) which was made from 2,2,2-trifluoroethylamine and 3,4-methylenedioxyphenylacetone and sodium cyanoborohydride in methanol. The best final line for this compound is that it is "possibly active." The most heroic dosage schedule mentioned was a total of 500 milligrams, taken in three approximately equal portions over the course of five or six hours, with only a very mild intoxication and little or no sympathomimetic effects. And what little there might have been was quickly gone. A collection of totally unexplored N-substituted homologues and analogues of MDE is gathered at the end of the recipe for MDBZ.
Another direction that has been used to homologate the MDMA and MDE structure is with the length of the aliphatic chain that carries the phenyl ring and the amine function. RHS shows the two-carbon chain, RIS shows the amphetamine chain length, and MDE can be called ETHYL-I. The four-carbon chain is the RJS group, and this entire Muni-Metro concept is explained under METHYL-J.
#107 MDHOET; HYDROXYETHYL-MDA; 3,4-METHYLENEDIOXY-N-(2-HYDROXYETHYL)AMPHETAMINE
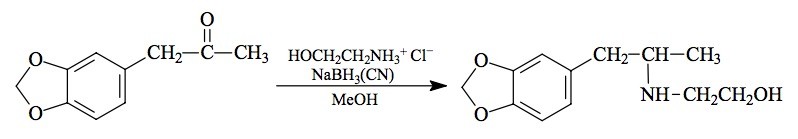

SYNTHESIS: To a well stirred solution of 25 g ethanolamine hydrochloride in 75 mL MeOH there was added 4.45 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 1.1 g sodium cyanoborohydride. Concentrated HCl in MeOH was added as required, over the next few days, to maintain the pH at about 6 as determined with external, dampened universal pH paper. The reaction mixture was added to 300 mL H2O and made strongly acidic with an excess of HCl. After washing with 3x100 mL CH2Cl2 the aqueous phase was made basic with 25% NaOH, and extracted with 4x100 mL CH2Cl2. Removal of the solvent under vacuum yielded 3.5 g of a viscous off-white oil that was distilled at 160 °C at 1.3 mm/Hg to give 2.0 g of a white viscous oil. The pot residue remained fluid, but was discarded. This distillate was dissolved in 8.0 mL IPA to give, eventually, a clear solution. This was neutralized with concentrated HCl and diluted with 100 mL anhydrous Et2O. The loose white crystals of 3,4-methylenedioxy-N-(2-hydroxy-ethyl)amphetamine hydrochloride (MDHOET) that formed were removed by filtration, washed with Et2O, and air dried. These weighed 2.3 g, and had a mp of 147-148 °C. Anal. (C12H18ClNO3) N.
DOSAGE: greater than 50 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: Most compounds with bare, exposed polar groups like hydroxyls are not centrally active, as they simply do not have any way of getting into the brain. MDHOET is certainly not very active, if it is active at all.
There was one report that at very high doses some central effects were indeed observed. With quantities in the several hundreds of milligrams a picture emerged of changes in perceived color and depth perception, but without euphoria. It was said to resemble a mild dose of ketamine. This is an interesting comment, in that ketamine has found its major medical use as an anesthetic, and MDHOET is among the most effective of all the N-substituted MDA derivatives assayed in several animal analgesia models.
#108 MDIP; N-ISOPROPYL-MDA; (3,4-METHYLENEDIOXY-N-ISOPROPYLAMPHETAMINE)
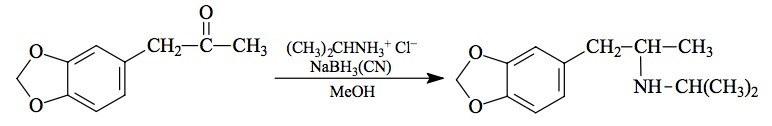

SYNTHESIS: To a well stirred and cooled solution of 14.75 g isopropylamine in 100 mL MeOH there was added 4.45 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by a 1:1 mixture of concentrated HCL and MeOH, sufficient to bring the pH to about 4. This was followed with 1.1 g sodium cyanoborohydride, and stirring was continued overnight. When the pH increased to over 6 there was added an additional 0.5 g of the borohydride, and additional methanolic HCl was added as needed to maintain the pH there. When the pH became stable, the reaction mixture was brought soundly acid with the addition of yet additional HCl, and all solvents were removed under vacuum. The residues were added to 500 mL H2O and washed with 3x100 mL CH2Cl2. The aqueous phase was made basic with 25% NaOH, and extracted with 4x100 mL CH2Cl2. Removal of the solvent under vacuum yielded 2.8 g of an amber liquid that was distilled at 95-110 °C at 0.3 mm/Hg. There was obtained about 2 mL of a white oil that was dissolved in 10 mL of IPA, neutralized with about 20 drops of concentrated HCl producing spontaneous crystals. These were diluted with some 40 mL of anhydrous Et2O, removed by filtration, washed with Et2O, and then air dried. There was obtained 1.6 g of 3,4-methylenedioxy-N-isopropylamphetamine hydrochloride (MDIP) with a mp of 186-186.5 °C with prior sintering at 185 °C. Anal. (C13H20ClNO2) N.
DOSAGE: greater than 250 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 250 mg) At 35 minutes there was an extremely slight head disturbance which increased over the next few minutes. I would have missed it if there had been any sensory input at all. At the one hour point there was a slight physical malaise, but no 'open window' of any kind, either like MDMA or like LSD. At the most, this was a threshold, and in another half hour, I was completely baseline.
EXTENSIONS AND COMMENTARY: The structure of MDIP can be looked at as exactly that of MDE but with an additional methyl group (one carbon) hanging off the ethyl that is on the nitrogen. And with that slight additional weight, the activity has disappeared. On those occasions where research has shown a compound to be inactive, there has been some study made that could be called a "primer" experiment. Why not take advantage of the fact that an "inactive" compound might well be sitting in some receptor site in the brain without doing anything? Might its presence, wherever it might be, have some effect if only a person were to explore it in the correct way? Might it augment or interfere with the action of another compound? Many experiments of this kind have been performed, geared to milk additional information out of a new trial of a new material.
Here is an example of a primer experiment that involved MDIP. Some five hours following an inactive trial with 120 milligrams of MDIP (maybe a slight disturbance at one hour, nothing at two hours) a calibration dose of 80 milligrams of MDMA was taken. The effects of the MDMA were noted at the 33 minute point, and an honest plus one was achieved at one hour. At this point a second 80 milligrams was added to the inventory that was already on board, and the general intoxication and the eye effects that followed were completely explained by the MDMA alone. It was obvious that the two drugs did not see one-another.
Sometimes an experiment can involve the assay of an unknown material at the supplement time of an active drug. This has been called "piggybacking." Here is an example. At the five hour point of an experiment with 140 milligrams of MDE (this had been a light experience, a plus one which had not laster more than two hours) a dosage of 200 milligrams of MDIP rekindled a +1 experience, a pleasant intoxication of the MDE sort, but one that was quite invested with tremor and some feelings of eye-popping. It was almost as if the physical toxic effects outweighed the mental virtues. Imagine an iceberg, with the bulk of its mass underwater. The MDE had had its own modest effects, and had submerged into invisibility, and the response to a little bit of an otherwise inactive MDIP was to refloat a bit of the otherwise unseeable MDE.
#109 MDMA; MDM; ADAM; ECSTASY; 3,4-METHYLENEDIOXY-N-METHYLAMPHETAMINE
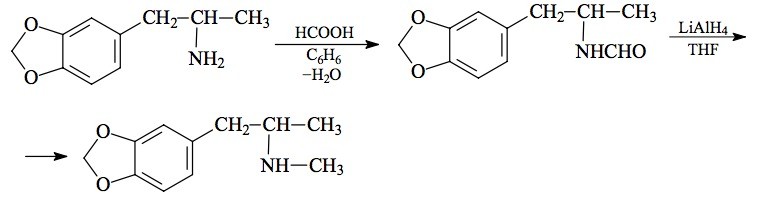

SYNTHESIS: (from MDA) A solution of 6.55 g of 3,4-methylenedioxyamphetamine (MDA) as the free base and 2.8 mL formic acid in 150 mL benzene was held at reflux under a Dean Stark trap until no further H2O was generated (about 20 h was sufficient, and 1.4 mL H2O was collected). Removal of the solvent gave an 8.8 g of an amber oil which was dissolved in 100 mL CH2Cl2, washed first with dilute HCl, then with dilute NaOH, and finally once again with dilute acid. The solvent was removed under vacuum giving 7.7 g of an amber oil that, on standing, formed crystals of N-formyl-3,4-methylenedioxyamphetamine. An alternate process for the synthesis of this amide involved holding at reflux for 16 h a solution of 10 g of MDA as the free base in 20 mL fresh ethyl formate. Removal of the volatiles yielded an oil that set up to white crystals, weighing 7.8 g.
A solution of 7.7 g N-formyl-3,4-methylenedioxyamphetamine in 25 mL anhydrous THF was added dropwise to a well stirred and refluxing solution of 7.4 g LAH in 600 mL anhydrous THF under an inert atmosphere. The reaction mixture was held at reflux for 4 days. After being brought to room temperature, the excess hydride was destroyed with 7.4 mL H2O in an equal volume of THF, followed by 7.4 mL of 15% NaOH and then another 22 mL H2O. The solids were removed by filtration, and the filter cake washed with additional THF. The combined filtrate and washes were stripped of solvent under vacuum, and the residue dissolved in 200 mL CH2Cl2. This solution was extracted with 3x100 mL dilute HCl, and these extracts pooled and made basic with 25% NaOH. Extraction with 3x75 mL CH2Cl2 removed the product, and the pooled extracts were stripped of solvent under vacuum. There was obtained 6.5 g of a nearly white residue which was distilled at 100-110 °C at 0.4 mm/Hg to give 5.0 g of a colorless oil. This was dissolved in 25 mL IPA, neutralized with concentrated HCl, followed by the addition of sufficient anhydrous Et2O to produce a lasting turbidity. On continued stirring, there was the deposition of fine white crystals of 3,4-methylenedioxy-N-methylamphetamine hydrochloride (MDMA) which were removed by filtration, washed with Et2O, and air dried, giving a final weight of 4.8 g.
(from 3,4-methylenedioxyphenylacetone) This key intermediate to all of the MD-series can be made from either isosafrole, or from piperonal via 1-(3,4-methylenedioxyphenyl)-2-nitropropene. To a well stirred solution of 34 g of 30% hydrogen peroxide in 150 g 80% formic acid there was added, dropwise, a solution of 32.4 g isosafrole in 120 mL acetone at a rate that kept the reaction mixture from exceeding 40 °C. This required a bit over 1 h, and external cooling was used as necessary. Stirring was continued for 16 h, and care was taken that the slow exothermic reaction did not cause excess heating. An external bath with running water worked well. During this time the solution progressed from an orange color to a deep red. All volatile components were removed under vacuum which yielded some 60 g of a very deep red residue. This was dissolved in 60 mL of MeOH, treated with 360 mL of 15% H2SO4, and heated for 3 h on the steam bath. After cooling, the reaction mixture was extracted with 3x75 mL Et2O, the pooled extracts washed first with H2O and then with dilute NaOH, and the solvent removed under vacuum The residue was distilled (at 2.0 mm/108-112 °C, or at about 160 °C at the water pump) to provide 20.6 g of 3,4-methylenedioxyphenylacetone as a pale yellow oil. The oxime (from hydroxylamine) had a mp of 85-88 °C. The semicarbazone had a mp of 162-163 °C.
An alternate synthesis of 3,4-methylenedioxyphenylacetone starts originally from piperonal. A suspension of 32 g electrolytic iron in 140 mL glacial acetic acid was gradually warmed on the steam bath. When quite hot but not yet with any white salts apparent, there was added, a bit at a time, a solution of 10.0 g of 1-(3,4-methylenedioxyphenyl)-2-nitropropene in 75 mL acetic acid (see the synthesis of MDA for the preparation of this nitrostyrene intermediate from piperonal and nitroethane). This addition was conducted at a rate that permitted a vigorous reaction free from excessive frothing. The orange color of the reaction mixture became very reddish with the formation of white salts and a dark crust. After the addition was complete, the heating was continued for an additional 1.5 h during which time the body of the reaction mixture became quite white with the product appeared as a black oil climbing the sides of the beaker. This mixture was added to 2 L H2O, extracted with 3x100 mL CH2Cl2, and the pooled extracts washed with several portions of dilute NaOH. After the removal of the solvent under vacuum, the residue was distilled at reduced pressure (see above) to provide 8.0 g of 3,4-methylenedioxyphenylacetone as a pale yellow oil.
To 40 g of thin aluminum foil cut in 1 inch squares (in a 2 L wide mouth Erlenmeyer flask) there was added 1400 mL H2O containing 1 g mercuric chloride. Amalgamation was allowed to proceed until there was the evolution of fine bubbles, the formation of a light grey precipitate, and the appearance of occasional silvery spots on the surface of the aluminum. This takes between 15 and 30 min depending on the freshness of the surfaces, the temperature of the H2O, and the thickness of the aluminum foil. (Aluminum foil thickness varies from country to country.) The H2O was removed by decantation, and the aluminum was washed with 2x1400 mL of fresh H2O. The residual H2O from the final washing was removed as thoroughly as possible by shaking, and there was added, in succession and with swirling, 60 g methylamine hydrochloride dissolved in 60 mL warm H2O, 180 mL IPA, 145 mL 25% NaOH, 53 g 3,4-methylenedioxyphenylacetone, and finally 350 mL IPA. If the available form of methylamine is the aqueous solution of the free base, the following sequence can be substituted: add, in succession, 76 mL 40% aqueous methylamine, 180 mL IPA, a suspension of 50 g NaCl in 140 mL H2O that contains 25 mL 25% NaOH, 53 g 3,4-methylenedioxyphenylacetone, and finally 350 mL IPA. The exothermic reaction was kept below 60 °C with occasional immersion into cold water and, when it was thermally stable, it was allowed to stand until it had returned to room temperature with all the insolubles settled to the bottom as a grey sludge. The clear yellow overhead was decanted and the sludge removed by filtration and washed with MeOH. The combined decantation, mother liquors and washes, were stripped of solvent under vacuum, the residue suspended in 2400 ml of H2O, and sufficient HCl added to make the phase distinctly acidic. This was then washed with 3x75 mL CH2Cl2, made basic with 25% NaOH, and extracted with 3x100 mL of CH2Cl2. After removal of the solvent from the combined extracts, there remained 55 g of an amber oil which was distilled at 100-110 °C at 0.4 mm/Hg producing 41 g of an off-white liquid. This was dissolved in 200 mL IPA, neutralized with about 17 mL of concentrated HCl, and then treated with 400 mL anhydrous Et2O. After filtering off the white crystals, washing with an IPA/Et2O mixture, (2:1), with Et2O, and final air drying, there was obtained 42.0 g of 3,4-methylenedioxy-N-methylamphetamine (MDMA) as a fine white crystal. The actual form that the final salt takes depends upon the temperature and concentration at the moment of the initial crystallization. It can be anhydrous, or it can be any of several hydrated forms. Only the anhydrous form has a sharp mp; the published reports describe all possible one degree melting point values over the range from 148-153 °C. The variously hydrated polymorphs have distinct infrared spectra, but have broad mps that depend on the rate of heating.
DOSAGE: 80 - 150 mg.
DURATION: 4 - 6 h.
QUALITATIVE COMMENTS: (with 100 mg) MDMA intrigued me because everyone I asked, who had used it, answered the question, 'What's it like?' in the same way: 'I don't know.' 'What happened?' 'Nothing.' And now I understand those answers. I too think nothing happened. But something seemed changed. Before the 'window' opened completely, I had some somatic effects, a tingling sensation in the fingers and temples Q a pleasant sensation, not distracting. However, just after that there was a slight nausea and dizziness similar to a little too much alcohol. All these details disappeared as I walked outside. My mood was light, happy, but with an underlying conviction that something significant was about to happen. There was a change in perspective both in the near visual field and in the distance. My usually poor vision was sharpened. I saw details in the distance that I could not normally see. After the peak experience had passed, my major state was one of deep relaxation. I felt that I could talk about deep or personal subjects with special clarity, and I experienced some of the feeling one has after the second martini, that one is discoursing brilliantly and with particularly acute analytical powers.
(with 100 mg) Beforehand, I was aware of a dull, uncaring tiredness that might have reflected too little sleep, and I took a modest level of MDMA to see if it might serve me as a stimulant. I napped for a half hour or so, and woke up definitely not improved. The feeling of insufficient energy and lack of spark that I'd felt before had become something quite strong, and might be characterized as a firm feeling of negativity about everything that had to be done and everything I had been looking forward to. So I set about my several tasks with no pleasure or enjoyment and I hummed a little tune to myself during these activities which had words that went: 'I shouldn't have done that, oh yes, I shouldn't have done that, oh no, I shouldn't have done that; it was a mistake.' Then I would start over again from the beginning. I was stuck in a gray space for quite a while, and there was nothing to do but keep doing what I had to do. After about 6 hours, I could see the whole mental state disintegrating and my pleasant feelings were coming back. But so was my plain, ornery tiredness. MDMA does not work like Dexedrine.
(with 120 mg) I feel absolutely clean inside, and there is nothing but pure euphoria. I have never felt so great, or believed this to be possible. The cleanliness, clarity, and marvelous feeling of solid inner strength continued throughout the rest of the day, and evening, and through the next day. I am overcome by the profundity of the experience, and how much more powerful it was than previous experiences, for no apparent reason, other than a continually improving state of being. All the next day I felt like 'a citizen of the universe' rather than a citizen of the planet, completely disconnecting time and flowing easily from one activity to the next.
(with 120 mg) As the material came on I felt that I was being enveloped, and my attention had to be directed to it. I became quite fearful, and my face felt cold and ashen. I felt that I wanted to go back, but I knew there was no turning back. Then the fear started to leave me, and I could try taking little baby steps, like taking first steps after being reborn. The woodpile is so beautiful, about all the joy and beauty that I can stand. I am afraid to turn around and face the mountains, for fear they will overpower me. But I did look, and I am astounded. Everyone must get to experience a profound state like this. I feel totally peaceful. I have lived all my life to get here, and I feel I have come home. I am complete.
(with 100 mg of the RRS isomer) There were the slightest of effects noted at about an hour (a couple of paresthetic twinges) and then nothing at all.
(with 160 mg of the RRS isomer) A disturbance of baseline at about forty minutes and this lasts for about another hour. Everything is clear by the third hour.
(with 200 mg of the RRS isomer) A progression from an alert at thirty minutes to a soft and light intoxication that did not persist. This was a modest +, and I was at baseline in another hour.
(with 60 mg of the RSS isomer) The effects began developing in a smooth, friendly way at about a half-hour. My handwriting is OK but I am writing faster than usual. At the one hour point, I am quite certain that I could not drive, time is slowing down a bit, but I am mentally very active. My pupils are considerably dilated. The dropping is evident at two hours, and complete by the third hour. All afternoon I am peaceful and relaxed, but clear and alert, with no trace of physical residue at all. A very successful ++.
(with 100 mg of the RSS isomer) I feel the onset is slower than with the racemate. Physically, I am excited, and my pulse and blood pressure are quite elevated. This does not have the 'fire' of the racemate, nor the rush of the development in getting to the plateau.
(with 120 mg of the RSS isomer) A rapid development, and both writing and typing are impossible before the end of the first hour. Lying down with eyes closed eliminates all effects; the visual process is needed for any awareness of the drug's effects. Some teeth clenching, but no nystagmus. Excellent sleep in the evening.
EXTENSIONS AND COMMENTARY: In clinical use, largely in psychotherapeutic sessions of which there were many in the early years of MDMA study, it became a common procedure to provide a supplemental dosage of the drug at about the one and a half hour point of the session. This supplement, characteristically 40 milligrams following an initial 120 milligrams, would extend the expected effects for about an additional hour, with only a modest exacerbation of the usual physical side-effects, namely, teeth clenching and eye twitching. A second supplement (as, for instance, a second 40 milligrams at the two and a half hour point) was rarely felt to be warranted. There are, more often than not, reports of tiredness and lethargy on the day following the use of MDMA, and this factor should be considered in the planning of clinical sessions.
With MDMA, the usual assignments of activity to optical isomers is reversed from all of the known psychedelic drugs. The more potent isomer is the RSS isomer, which is the more potent form of amphetamine and methamphetamine. This was one of the first clear distinctions that was apparent between MDMA and the structurally related psychedelics (where the RRS isomers are the more active). Tolerance studies also support differences in mechanisms of action. In one study, MDMA was consumed at 9:00 AM each day for almost a week (120 milligrams the first day and 160 milligrams each subsequent day) and by the fifth day there were no effects from the drug except for some mydriasis. And even this appeared to be lost on the sixth day. At this point of total tolerance, there was consumed (on day #7, at 9:00 AM) 120 milligrams of MDA and the response to it was substantially normal with proper chronology, teeth clench, and at most only a slight decrease in mental change. A complete holiday from any drug for another 6 days led to the reversal of this tolerance, in that 120 milligrams of MDMA had substantially the full expected effects. The fact that MDMA and MDA are not cross-tolerant strengthens the argument that they act in different ways, and at different sites in the brain.
A wide popularization of the social use of MDMA occurred in 1984-1985 and, with the reported observation of serotonin nerve changes in animal models resulting from the administration of the structurally similar drug MDA, an administrative move was launched to place it under legal control. The placement of MDMA into the most restrictive category of the Federal Controlled Substances Act has effectively removed it from the area of clinical experimentation and human research. The medical potential of this material will probably have to be developed through studies overseas.
A word of caution is in order concerning the intermediate 3,4-methylene-dioxyphenylacetone, which has also been called piperonylacetone. A devilish ambiguity appeared in the commercial market for this compound, centered about its name. The controversy focused on the meaning of the prefix, piperonyl, which has two separate chemical definitions. Let me try to explain this fascinating chaos in non-chemical terms. Piperonyl is a term that has been used for a two-ring system (the methylenedioxyphenyl group) either without, or with, an extra carbon atom sticking off of the side of it. Thus, piperonylacetone can be piperonyl (the two-ring thing without the extra carbon atom attached) plus acetone (a three carbon chain thing); the total number of carbons sticking out, three. Or, piperonylacetone can be piperonyl (the two-ring thing but with the extra carbon atom attached) plus acetone (a three carbon chain thing); the total number of carbons sticking out, four.
Does this make sense?
The three carbon sticking out job gives rise to MDA and to MDMA and to many homologues that are interesting materials discussed at length in these Book II comments. This is the usual item of commerce, available from both domestic and foreign suppliers. But the four-carbon sticking out job will produce totally weird stuff without any apparent relationship to psychedelics, psychoactives or psychotropics whatsoever. I know of one chemical supply house which supplied the weird compound, and they never did acknowledge their unusual use of the term piperonyl. There is a simple difference of properties which might be of value. The three carbon (correct) ketone is an oil with a sassafras smell that is always yellow colored. The four carbon (incorrect) ketone has a weak terpene smell and is white and crystalline. There should be no difficulties in distinguishing these two compounds. But unprincipled charlatans can always add mineral oil and butter yellow to otherwise white solids to make them into yellow oils. Caveat emptor.
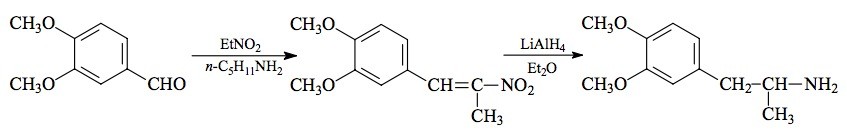
#110 MDMC; EDMA; 3,4-ETHYLENEDIOXY-N-METHYLAMPHETAMINE
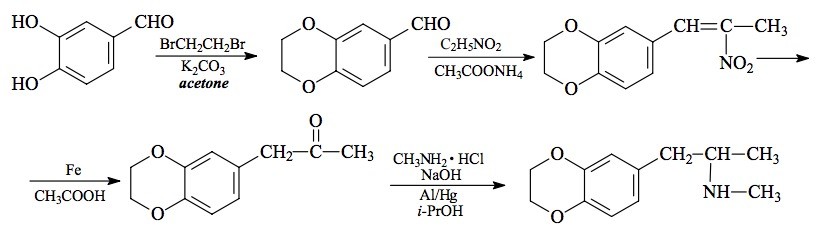

SYNTHESIS: To a solution of 27.6 g protocatechualdehyde (3,4-dihydroxybenzaldehyde) in 250 mL acetone there was added 57 g finely powdered anhydrous K2CO3 and 43 g 1,2-dibromoethane. The mixture was held at reflux for 16 h, and then the acetone removed by evaporation. The remaining tar-like goo was distributed between equal volumes of H2O and CH2Cl2, and the phases separated by centrifugation. The organic phase was washed with 2x50 mL 5% NaOH, and the solvent removed under vacuum. The residue (22.0 g with the smell of the starting halide) was distilled to give a fraction that boiled at 110 °C at 0.25 mm/Hg to yield 3,4-ethylenedioxybenzaldehyde (1,4-benzodioxane-6-carboxaldehyde) as a white oil weighing 6.88 g. This spontaneously crystallized to give white solids that melted at 50-51 °C.
A solution of 6.64 g 3,4-ethylenedioxybenzaldehyde in 40 mL nitroethane was treated with 0.26 g anhydrous ammonium acetate and held at reflux for 3 days. TLC analysis showed that there was much aldehyde remaining unreacted, so an additional 0.7 g ammonium acetate was added, and the mixture held at reflux for an additional 6 h. The excess nitroethane was removed under vacuum. The residue was dissolved in 30 mL hot MeOH which, with patience and slow cooling, finally deposited a heavy yellow-gold powder. This product 1-(3,4-ethylenedioxyphenyl)-2-nitro-propene melted at 95-96 °C and weighed 6.03 g when air dried to constant weight. Recrystallization from either MeOH or EtOAc gave the product as a yellow solid, but without any improvement in mp.
A solution of 4.0 g of 1-(3,4-ethylenedioxyphenyl)-2-nitropropene was made in 30 mL warm acetic acid. This was added to a suspension of 16 g elemental electrolytic iron in 75 mL acetic acid. The mixture was heated on the steam bath, and an exothermic reaction set in at about 70 °C. Heating was continued and the reaction allowed to proceed until the mass was a thick gray color and a dirty scum had been formed on the surface. After about 2 h, the entire mix was poured into 2 L H2O and filtered free of a little residual unreacted iron which was washed with CH2Cl2. The filtrate and washes were extracted with 3x100 mL CH2Cl2 and the pooled organic extracts washed with 2x50 mL 5% NaOH. Removal of the solvent gave 3.38 g of an amber oil which was distilled. The product 1-(3,4-ethylenedioxyphenyl)-2-propanone distilled as a white oil, at 105-110 °C at 0.2 mm/Hg. It weighed 2.74 g.
To 2.0 g. of 1 inch squares of light-weight aluminum foil there was added a solution of 50 mg mercuric chloride in 70 mL water. After standing at room temperature for 30 min, the H2O was drained away, and the amalgamated aluminum washed twice with H2O, and shaken as dry as possible. There was then added, promptly and in immediate sequence, a solution of 3 g methylamine hydrochloride in 3 mL H2O, 9 mL IPA, 7.25 mL 25% NaOH, 2.70 g of 1-(3,4-ethylenedioxyphenyl)-2-propanone, and 18 mL IPA. The mixture was heated on the steam bath until an exothermic reaction set in, and then it was continuously swirled as the reaction proceeded. When the aluminum was consumed, there was a colorless gray sludge, and this was filtered and washed with 2x10 mL MeOH. The combined mother liquors and washes were stripped of solvent under vacuum. The two phase residue was suspended in 400 mL H2O containing sufficient H2SO4 to make the resulting water solution acidic to pH paper. This was washed with 3x50 mL CH2Cl2, made basic with 25% NaOH, and the product extracted with 3x50 mL CH2Cl2. The resulting 3.01 g slightly amber residue oil was distilled at 110-120 °C at 0.25 mm/Hg to give 2.53 g of a white oil, which did not appear to absorb carbon dioxide. This was dissolved in 12 mL IPA, neutralized with 1 mL concentrated HCl and diluted with anhydrous Et2O to the point of initial turbidity. There separated white crystals of 3,4-ethylenedioxy-N-methylamphetamine hydrochloride (MDMC) which weighed, when air dried to constant weight, 2.53 g.
DOSAGE: 200 or more mg.
DURATION: 3 - 5 h.
QUALITATIVE COMMENTS: (with 150 mg) A flood of paresthesia at the 30 minute point, and then nothing. There was the development of a plus one-and-a half effect over the next hour with the tendency to drift into a dozing state with hypnogogic imagery. There were colored letters in the periphery of my visual field. There was no appetite loss nor was there any blood pressure rise. And no eye jiggle or teeth clenching. I was out of the experience in 4 to 5 hours. A repeat of this level a few days later gave a bare possible threshold with no other effects.
(with 200 mg) There was something unmistakable at 45 minutes, with hints of nystagmus. Possibly MDMA-like, with no indicators of anything psychedelic. Subtle return to baseline, and there were no after-effects.
(with 250 mg) Alert at 40 minutes, and to a clear ++ at an hour. Slight something in the eye muscles. Dropping thirty minutes later, and baseline at three hours.
(with 250 mg) I am at a bare threshold at best.
EXTENSIONS AND COMMENTARY: What a strange and completely unsatisfactory compound° In the original run-up from low levels to increasing higher levels, there never was a dosage that was a minus, that had no effect. At every level, something was thought to be there, usually at a level of a single plus or thereabouts. But with different people, different responses. There is no way of guessing what an active level might be, or how consistent that level might be between different people, or for that matter what the responses are that might be expected at that level.
This was yet one more effort to find an MDMA-like substitute by the miniscule manipulation of the MDMA molecule. Perhaps a small molecular change might leave the particular magic of the MDMA action alone, but eliminate the serotonin neuron problem in test animals. Maybe the serotonin neuron change is essential for MDMA to have the action it has. Who can tell?
The original name that this compound got, during the several explorations of MDMA analogues, was based on the nickname for MDMA which was Adam. HADUEM was mentioned with the hydroxy compound, MADAM with the 6-methyl homologue, and FLADAM with the 6-fluoro analogue. This compound got the sobriquet MACADAM from that horrible black gooey mess generated at the aldehyde stage. This was shortened to RCS and eventually the RCS was added to the MDMA parent name. Thus, MDMC. It doesn't really make sense; EDMA is more reasonable. But then there is no reason why MDMC should make sense.
#111 MDMEO; N-METHOXY-MDA; 3,4-METHYLENEDIOXY-N-METHYOXYAMPHETAMINE
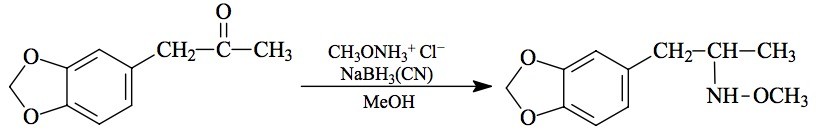

SYNTHESIS: To a solution of 20.9 g methoxyamine hydrochloride in 75 mL MeOH (a strongly acidic solution) there was added 4.45 g 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 1.10 g sodium cyanoborohydride. There was the immediate formation of a solid phase, and the evolution of what appeared to be hydrogen cyanide. To this there were added about 4 mL 5% NaOH which brought the pH to the vicinity of 3 or 4. Another 1.0 g of sodium cyanoborohydride was added (no gas evolution this time) and stirring was continued at ambient temperature for 6 days. All was added to 500 mL H2O, acidified with 10 mL HCl, and extraction with 3x100 mL CH2Cl2 removed almost all the color. The aqueous phase was made basic with 25% NaOH, and extracted with 4x100 mL CH2Cl2. Evaporation of the solvent from these extracts yielded 1.8 g of a pale yellow oil which, on distillation at 90-95 °C at 0.5 mm/Hg, gave a 1.6 g fraction of an absolutely white, viscous, clear oil. This was dissolved in 8 mL IPA and neutralized with concentrated HCl. The product was an exceptionally weak base, and appropriate end points must be respected on the external pH paper (yellow to red, rather than purple to orange). Anhydrous Et2O was added to the point of turbidity, and as soon as crystallization had actually started, more Et2O was added with stirring, for a net total of 200 mL. After a couple of h standing, the fine white crystalline 3,4-methylenedioxy-N-methoxyamphetamine hydrochloride (MDMEO) was removed by filtration, Et2O washed, and air dried to constant weight. There was obtained 1.7 g of a product with a mp of 143-146 °C. The proton NMR was excellent with the N-methoxyl group a sharp singlet at 4.06 ppm. Anal. (C11H16ClNO3) N.
DOSAGE: greater than 180 mgs.
DURATION: unknown
EXTENSIONS AND COMMENTARY: Why the interest in the N-methoxy analogue of MDA? There are several reasons. One, this is an isostere of MDE and it would be interesting to see if it might serve as a primer to the promotion of the effectiveness of other drugs (see primer discussion under MDPR). In one experiment, wherein a 60 microgram dosage of LSD was used an hour and a half after a 180 milligram load of MDMEO, there was no augmentation of effects. Thus, it would appear not to be a primer. Another reason for interest was that the material, although having an extremely similar overall structure to most of the active MD-series compounds, is very much a weaker base. And MDOH, which is also a very much weaker base than MDA, still shows the action and potency of MDA. And, as this compound appears to be inactive, base strength is not a sole predictor of activity.
The ultimate reason for making MDMEO was, of course, that it could be made. That reason is totally sufficient all by itself.
#112 MDMEOET; N-METHOXYETHYL-MDA; 3,4-METHYLENEDIOXY-N-(2-METHOXYETHYL)AMPHETAMINE
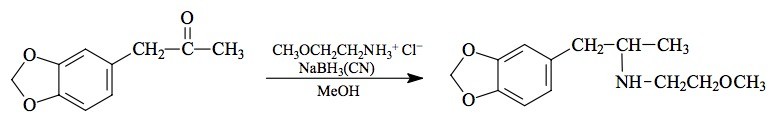

SYNTHESIS: A crude solution of methoxyethylamine hydrochloride was prepared from 17.7 g methoxyethylamine and 20 mL concentrated HCl with all volatiles removed under vacuum. This was dissolved in 75 mL MeOH and there was added 4.45 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 1.3 g sodium cyanoborohydride. Concentrated HCl in MeOH was added as required to maintain the pH at about 6 as determined with external, dampened universal pH paper. About 4.5 mL were added over the course of 5 days, at which time the pH had stabilized. The reaction mixture was added to 400 mL H2O and made strongly acidic with an excess of HCl. After washing with 2x100 mL CH2Cl2 the aqueous phase was made basic with 25% NaOH, and extracted with 4x75 mL CH2Cl2. Removal of the solvent under vacuum yielded 6.0 g of an amber oil that was distilled at 110-120 °C at 0.2 mm/Hg. There was obtained 4.7 g of a crystal-clear white oil that was dissolved in 30 mL IPA and neutralized with 45 drops of concentrated HCl producing a heavy mass of spontaneous crystals that had to be further diluted with IPA just to be stirred with a glass rod. These were diluted with 200 mL of anhydrous Et2O, removed by filtration, and washed with additional Et2O. After air drying there was obtained 4.9 g of 3,4-methylenedioxy-N-(2-methoxyethyl)amphetamine hydrochloride (MDMEOET) with a mp of 182.5-183 °C. Anal. (C13H20ClNO3) N.
DOSAGE: greater than 180 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: This is another example of the replacement of a neutral atom out near the end of a chain, with a more basic and a more polar one. MDMEOET would be called an isostere of MDBU in that it has the same shape, with a methylene unit (the CH2) replaced by an oxygen atom. No activity turned up with either compound, so nothing can be learned from this particular example of change of polarity.
#113 MDMP; a,a,N-TRIMETHYL-3,4-METHYLENEDIOXY-PHENETHYLAMINE; METHYLENEDIOXYMEPHENTERMINE
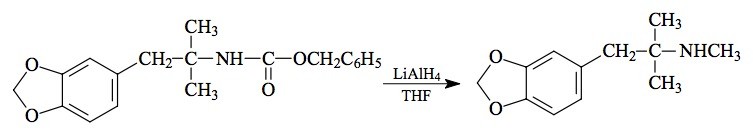

SYNTHESIS: To a well stirred solution of 1.64 g of 1-(N-(benzyloxycarbonyl)amino)-1,1-dimethyl-2-(3,4-methylenedioxyphenyl)ethane (see under MDPH for its preparation) in 10 mL anhydrous THF there was added a suspension of 0.38 g LAH in 25 mL THF. All was held at reflux for 24 h, the excess hydride was destroyed by the addition of 1.5 mL H2O, and sufficient aqueous NaOH was added to make the reaction mixture basic and flocculant enough to be filterable. The inorganic solids were removed by filtration and, following washing with THF, the combined filtrate and washings were stripped of organic solvent under vacuum. The residue was dissolved in 100 mL Et2O and washed with 2x50 mL saturated aqueous NaHCO3. After drying the organic phase with anhydrous MgSO4, the solvent was removed under vacuum to give a yellow oil. This was dissolved in 50 mL absolute EtOH and neutralized with concentrated HCl. Removal of the solvent under vacuum yielded an off-white solid that was recrystallized from an EtOH/EtOAc mixture to provide 0.84 g of a,a,N-trimethyl-3,4-methylenedioxyphenethylamine hydrochloride (MDMP) with a mp of 206-208 °C. The NMR spectrum showed the a,a-dimethyl pair as a singlet at 1.38 ppm. Anal. (C12H18ClNO2) C,H,N.
DOSAGE: above 110 mg.
DURATION: perhaps 6 hours.
QUALITATIVE COMMENTS: (with 60 mg) There was a faint, dull alerting at just over a half hour. The time sense was out of order, and an absence of visuals but a generalized attentiveness to my surroundings was suggestive of MDMA. Nothing remained at the six hour point.
(with 110 mg) There was a light-headedness, and a complete absence of libido. Nothing in any way psychedelic, but there are hints of discomfort (jaw tension) that will bear close watching at higher dosages. It might evolve at higher levels into something like MDMA.
EXTENSIONS AND COMMENTARY: This is one of several candidates for clinical use as a substitute for MDMA, but there will have to be a much broader study of its qualitative action in man. It is clearly not psychedelic at these modest levels, and in in vitro animal studies it was apparently inactive as a serotonin releaser. The warped logic for looking at phentermine analogs was discussed in the comments that concerned MDPH. The initials used here have been chosen with care. MDM should not be used as it has found some currency as an abbreviation for MDMA (Methylene-Dioxy-Methamphetamine). MDMP fits neatly with Methylene-Dioxy-Me-Phentermine.
#114 MDOH; N-HYDROXY-MDA; 3,4-METHYLENEDIOXY-N-HYDROXYAMPHETAMINE
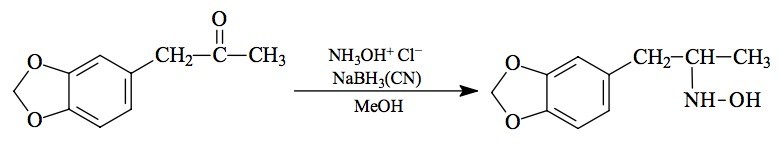

SYNTHESIS: To a well stirred solution of 14.8 g hydroxylamine hydrochloride in 120 mL MeOH there was added 3.6 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 1.0 g sodium cyanoborohydride. The oxime, prepared from the ketone and hydroxylamine in MeOH with pyridine, may be substituted for these two components. Concentrated HCl was added over the course of a couple of days, to keep the pH near neutrality. When the reaction was complete, it was added to H2O, made strongly acidic with HCl, and washed with 3x100 mL CH2Cl2. The aqueous phase was made basic with 25% NaOH, and reextracted with 3x100 mL of CH2Cl2. The extracts were pooled, and the solvent removed under vacuum to give 1.7 g of an oily residue which, with pumping under a hard vacuum for a few minutes, changed to a white solid. This can be Kugelrohred if the vacuum is sufficiently good to keep the temperature during the distillation below 100 °C. The extremely viscous distillate formed crystals immediately upon wetting with IPA. It was dissolved in 20 mL of warm IPA and neutralized with concentrated HCl, with the titration end-point being red rather than orange on universal pH paper. Modest addition of Et2O allowed the formation of 3,4-methylenedioxy-N-hydroxyamphetamine hydrochloride (MDOH) as white crystals, which weighed 1.4 g when air dried. If the temperature of distillation exceeded 100 °C, there was extensive decomposition during distillation, with the formation of 3,4-methylenedioxyamphetamine (MDA) and the oxime of the ketone. Under these circumstances, the only base isolated was MDA. The surest isolation procedure was to obtain MDOH as the free base, as a crystalline solid which could be recrystallized from 5 volumes of boiling IPA. The free base had a mp of 94-95 °C (and should not be confused with the oxime of 3,4-methylenedioxyphenylacetone which has a mp of 86-88 °C since the mixed mp is depressed, mp 56-62 °C, or with the free base of MDA which is an oil). Anal. (C10H13NO3) N. The hydrochloride salt had a mp of 149-150 °C (and should not be confused with the hydrochloride of MDA which has a mp of 185-186 °C since the mixed mp is depressed, mp 128-138 °C). Anal. (C10H14ClNO3) N. Acetic anhydride can serve as a useful tool for distinguishing these materials. MDA gives an N-acetyl derivative with an mp of 92-93 °C. MDOH gives an N,O-diacetyl derivative with a mp of 72-74 °C. Methylenedioxyphenylacetone oxime gives an O-acetyl derivative that is an oil.
DOSAGE: 100 - 160 mg.
DURATION: 3 - 6 h.
QUALITATIVE COMMENTS: (with 100 mg) I felt hampered the first hour by some internal barrier, which prevented total enjoyment. However, this began to break through in a wonderful way just before the supplement was offered. Since I felt I was beginning to move through the barrier, I declined the supplement, particularly since I was anxious to compare the after-effects with my first experience. I had found the first time very remarkable, but felt unusually tired for several days following. I feel it is important to know whether this is a specific drug-induced effect, or the result of psychological phenomena. The experience continued in a rich, meaningful way. There was a marvelous inner glow, the warmth from all the other participants was wonderful to feel, nature was most beautiful. There were no dramatic breakthroughs, or rushes of insight or energy, but just a wonderful contemplative space where things gently unfolded as you put your attention on them.
(with 100 mg) The material came on fairly rapidly. In about 30 minutes, I was intensely intoxicated, and more deeply than with MDMA. It was a glorious feeling, and beauty was everywhere enhanced. With eyes closed it felt marvelous, and it was appealing to pursue the inner experience. I did notice an internal dryness which was characteristic of MDMA, and I had similar difficulty in urinating, but not as intense as with MDMA.
(with 120 mg) The colors of the market-place, of all the fresh foods, constituted a beautiful mosaic. Nothing practical, simply a real treasure to be used with individual intention and enjoyment. Everything was seen with new eyes, new meanings, faces, figures, the colors of the rainbow subconsciously individually applied. A 'soul-scape'. The following day very exhausted, tired, back-pain.
EXTENSIONS AND COMMENTARY: The first time that MDOH was synthesized, it had inadvertently and unknowingly been converted to MDA. And the search for proper dosage and characterization of effects of this product was, of course, the rediscovery of the dosage and the effects of MDA. It is one of the world's most remarkable coincidences that after the second synthesis of MDOH, when MDOH had really and truly been actually prepared, the brand new search for proper dosage and characterization of effects revealed that they were almost identical to the earlier observations for (the inadvertently produced) MDA.
This reminds me of my speculations in the discussion of both FLEA and the HOT compound where they also showed paired molecular structures with their prototypes that differ only by a single oxygen atom. Again, might there be some metabolic interconversion within the body? The immediate thought would be that the oxygen atom (the hydroxy group) might be metabolically removed, and the effects of either drug are due to the action of MDA. But the opposite direction is in many ways more appealing, the in vivo conversion of MDA to MDOH. Why more appealing? For one thing, oxidative changes are much more common in the body than reductive changes. For another, the conversion of amphetamine to N-hydroxyamphetamine is an intermediate in the conversion of amphetamine to phenylacetone, a known metabolic process in several animal species. And that intermediate, N-hydroxyamphetamine, is a material that gives the famous cytochrome P-450 complex that has fascinated biochemists studying the so-called NADPH-dependent metabolism.
I would put my money on the likelihood of MDA going to MDOH if it should turn out that the two drugs interconvert in the body. And in that case, it would be MDOH, or another metabolite on down the line that is common to both MDA and MDOH, that is the factor intrinsic to the intoxication that is produced. Human metabolic studies are needed, and they have not yet been done.
#115 MDPEA; 3,4-METHYLENEDIOXYPHENETHYLAMINE; HOMOPIPERONYLAMINE
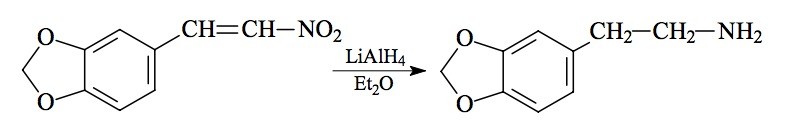

SYNTHESIS: A suspension of 4.0 g LAH in 300 mL anhydrous Et2O was stirred and heated to a gentle reflux in an inert atmosphere. There was added 3.9 g 3,4-methylenedioxy-'-nitrostyrene (see under BOH for its preparation) by allowing the condensing Et2O to leach it out from a Soxhlet thimble. After the addition was complete, the reaction mixture was held at reflux for an additional 48 h. It was then cooled and the excess hydride was destroyed by the cautious addition of 300 mL of 1.5 N H2SO4. When both phases were completely clear, they were separated, and the aqueous phase washed once with 50 mL Et2O. There was then added 100 g potassium sodium tartrate, followed by sufficient base to bring the pH >9. This was extracted with 3x75 mL CH2Cl2, and the solvent from these pooled extracts was removed under vacuum. The residue was dissolved in 150 mL anhydrous Et2O and saturated with anhydrous HCl gas. There was a heavy crystallization of 3,4-methylenedioxyphenethylamine hydrochloride (MDPEA) which weighed 3.0 g and had a mp of 212-213 °C.
DOSAGE: greater than 300 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 200 mg) It was taken twice at different times in a dosage of 200 milligrams each time, without the slightest peripheral or central effects.
(with 300 mg) My tinnitus had disappeared. Probably nothing.
EXTENSIONS AND COMMENTARY: How strange. Even more than DMPEA, this cyclic analogue MDPEA is a potential prodrug to dopamine, and would be a prime candidate for central activity. So why is this drug not active? The usual reason advanced by the pharmacologists is that the body is full of potent enzymes known as monoamine oxidases, and this is a monoamine, and so the body simply chews away on it in an oxidative manner, inactivating it before it ever makes it to some target receptor.
That is the pitch given in the textbooks. Phenethylamines are subject to easy enzymatic oxidation, hence they are not active. The presence of an alpha-methyl group (the corresponding amphetamines) blocks the compound from easy access to the enzyme, and since that protects them from oxidative destruction, they are active. The oft-quoted exception is mescaline, and even it is largely destroyed, as evidenced by the large amount needed for activity (a fraction of a gram). Sorry, I can't buy it. This entire book is peppered with phenethylamines that are active at the few-milligram area. Why aren't they also destroyed as well? The textbooks simply are not right.
MDPEA was one of the seven compounds evaluated as to toxicity and animal behavior at the University of Michigan under contract from the Army Chemical Center. Its Edgewood Arsenal code number was EA-1297. The number for MDA itself was EA-1298.
The beta-hydroxy analogue of MDPEA is the ethanolamine MDE, standing for methylenedioxyethanolamine. This is an old term, and in the more recent literature, since 1975 certainly, MDE has been used to represent methylenedioxyethylamphetamine. The ethanolamine compound is discussed in the recipe for DME.
There is a family of compounds, to be discussed elsewhere, that is called the Muni-Metro (see under METHYL-J). The simplest member is this compound, MDPEA, and under its chemically acceptable synonym, homopiperonylamine, it can be called RHS. Following that code, then, the N-methyl homologue of MDPEA is METHYL-H, and it has been looked at, clinically, as an antitussive agent. N-METHYL-MDPEA, or METHYL-H, or N-methyl-3,4-methylenedioxyphenethylamine is effective in this role at dosages of about 30 milligrams, but I have read nothing that would suggest that there were any central effects. I have tried it at this level and have found a little tightness of the facial muscles, but there was nothing at all in the mental area.
#116 MDPH; a,a-DIMETHYL-3,4-METHYLENEDIOXY-PHENETHYLAMINE; 3,4-METHYLENEDIOXYPHENTERMINE
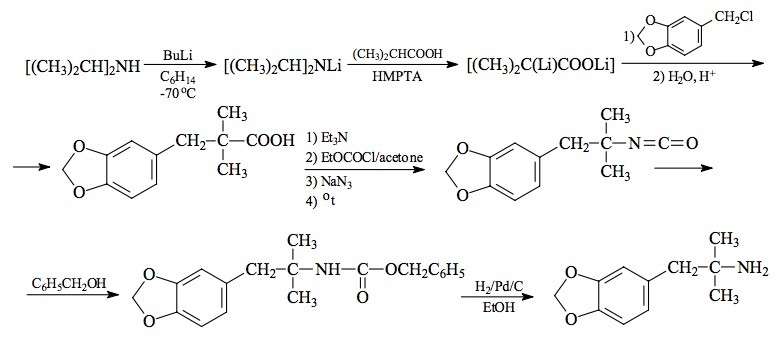

SYNTHESIS: To 150 mL of THF, under an atmosphere of nitrogen, there was added 11.2 g diisopropylamine, and the solution was cooled with external dry ice/IPA. There was then added 48 mL of a 2.3 M solution of butyllithium in hexane, dropwise, with good stirring. This was warmed to room temperature, stirred for a few min, and then all was cooled again in the dry ice bath. Following the dropwise addition of 4.4 g of isobutyric acid there was added 10.5 mL hexamethylphosphoramide. Again, the stirred reaction mixture was brought to room temperature for about 0.5 h. There was then added, drop-wise, 8.5 g 3,4-methylenedioxybenzyl chloride and the mixture allowed to stir overnight at room temperature. The reaction mixture was poured into 100 mL 10% HCl, and the excess THF was removed under vacuum. The acidic aqueous residue was extracted with 2x150 mL Et2O. These extracts were pooled, washed with 10% HCl, and then extracted with 3x75 mL of 4 N Na2CO3. These extracts were pooled, made acidic with HCl, and again extracted with Et2O. After drying the pooled extracts with anhydrous MgSO4, the solvent was removed under vacuum to give a residue that spontaneously crystallized. Recrystallization from hexane yielded 6.5 g of 2,2-dimethyl-3-(3,4-methylenedioxyphenyl)propionic acid as white crystals with a mp of 71-73 °C. The NMR spectrum in CDCl3 showed the alpha-dimethyl groups as a sharp singlet at 1.18 ppm. Anal. (C12H14O4) C,H.
The triethylamine salt of 2,2-dimethyl-3-(3,4-methylenedioxyphenyl)propionic acid (5.4 g amine, 11.4 g acid) was dissolved in 10 mL H2O and diluted with sufficient acetone to maintain a clear solution at ice-bath temperature. A solution of 6.4 g ethyl chloroformate in 40 mL acetone was added to the 0 °C solution over the course of 30 min, followed by the addition of a solution of 4.1 g sodium azide in 30 mL H2O. Stirring was continued for 45 min while the reaction returned to room temperature. The aqueous phase was extracted with 100 mL toluene which was washed once with H2O and then dried with anhydrous MgSO4. This organic solution of the azide was heated on a steam bath until nitrogen evolution had ceased, which required about 30 min. The solvent was removed under vacuum and the residue was dissolved in 30 mL benzyl alcohol. This solution was heated on the steam bath overnight. Removal of the excess benzyl alcohol under vacuum left a residue 13.5 g of 1-(N-(benzyloxycarbonyl)amino)-1,1-dimethyl-2-(3,4-methylenedioxyphenyl)ethane as an amber oil. The dimethyl group showed, in the NMR, a sharp singlet at 1.30 ppm in CDCH3. Anal. (C19H21NO4) C,H. This carbamate was reduced to the primary amine (below) or to the methylamine (see under MDMP).
A solution of 3.27 g of 1-[N-(benzyloxycarbonyl)amino]-1,1-dimethyl-2-(3,4-methylenedioxyphenyl)ethane in 250 mL absolute ethanol was treated with 0.5 g 10% palladium on carbon. This mixture was shaken under hydrogen at 35 pounds pressure for 24 h. The carbon was removed by filtration through Celite, and the filtrate titrated with HCl. The solvent was removed under vacuum, and the residue allowed to crystallize. This produce was recrystallized from an EtOH/EtOAc mixture to provide a,a-dimethyl-3,4-methylenedioxyphenethylamine hydrochloride (MDPH). The white crystals weighed 1.63 g and had a mp of 180-181 °C. Anal. (C11H16ClNO2) C,H,N.
DOSAGE: 160 - 240 mg.
DURATION: 3 - 5 h.
QUALITATIVE COMMENTS: (with 120 mg) The alert was felt in forty minutes and I was pretty much there at an hour and twenty. Quite like MDA, simple, with no lines, no colors, no motion, no fantasy. I am pleasantly stoned. The anorexia is real, as is the impotency. The drop from the 4th to the 6th hour was softened by a modest amount of wine, and this proved to be extremely intoxicating. My speech was slurred, and there was later amnesia for the rather aggressive and uninhibited behavior that occurred. I felt that there was more drug than alcohol contributing to this episode. My dream patterns were disturbingly unreal.
(with 160 mg) A very quiet development. There was no body load whatsoever. And no visual, and I saw it fading away all too soon. This might be a good promoter, like MDPR. I felt refreshed and relaxed on the following morning.
(with 200 mg) This has an inordinately foul taste. I felt slightly queasy. There were short daydreams which were quickly forgotten. I see no values that are worth the hints of physical problems, a little eye mismanagement and some clenching of teeth, and a tendency to sweat. I was able to sleep at only five hours into it, but there were a couple of darts. This is not as rewarding (stoning) as MDA, and has none of the magic of MDMA. It was a short-lived plus two.
EXTENSIONS AND COMMENTARY: What is the train of thought that leads from the structure of a known compound (which is active) to the structure of an unknown one (which may or may not be active)? Certainly the extrapolations involve many what-if's and maybe's. The path can be humorous, it certainly can be tortuous, and it often calls for special things such as faith, insight, and intuition. But can one say that it is logical?
Logic is a tricky thing to evaluate. One of the earliest approaches was laid down by Aristotle, in the form of the syllogism. In it there are three lines consisting of two premises and a conclusion, a form that is called a "mood." All are statements of relationships and, if the premises are true, there are only certain conclusions that may logically follow. For example:
- Every man is a lover.
- Every chemist is a man.
- Therefore, every chemist is a lover.
Letting lover be the major term RaS and letting chemist be the minor term RbS and letting man be the middle term RmS, this reduces to:
- Every m is a,
- Every b is m.
- Therefore, every b is a
and it is a valid mood called Barbara.
Of the 256 possible combinations of all's and some's and none's and are's and are-not's, only 24 moods are valid. The reasoning here with MDPH goes:
Some stimulants when given a methylenedioxy ring are MDMA-like.
Some ring-unsubstituted 1,1-dimethylphenylethylamines are stimulants.
Therefore, some ring-unsubstituted 1,1-dimethylphenylethylamines when given a methylenedioxy ring are MDMA-like.
In symbolic form this is:
- Some m is a, and
- Some b is m, then
- Some b is a
and this is not one of the 24 valid moods. Given the first premise as some m is a, there is only one valid syllogism form that can follow, and this is known as Disamis, or:
- Some m is a, and
- Every m is b, then
- Some b is a
which translates as:
Some stimulants when given a methylenedioxy group are MDMA-like.
Every stimulant is a ring-unsubstituted 1,1-dimethylphenylethylamine.
Therefore, some ring-unsubstituted 1,1-dimethylphenylethylamines when given a methylenedioxy group are MDMA-like.
The conclusion is the same. But the second premise is false so the entire reasoning is illogical. What is the false second premise? It is not a fact that every stimulant is a phentermine. There are lots of stimulants that are not phentermines.
So much for applying syllogistics to pharmacology.
#117 MDPL; N-PROPARGYL-MDA; N-PROPYNYL-MDA; 3,4-METHYLENEDIOXY-N-PROPARGYLAMPHETAMINE)
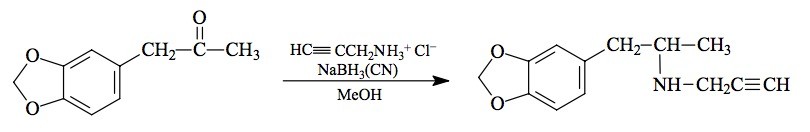

SYNTHESIS: A solution of 10.5 g propargylamine hydrochloride in 40 mL MeOH was treated with 2.0 g 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 0.55 g sodium cyanoborohydride. Concentrated HCl was added as needed, to keep the pH constant at about 6. The reaction seemed to progress very slowly. After about five days, the reaction mixture was added to 400 of H2O, acidified with HCl, and extracted with 3x100 mL CH2Cl2. The aqueous phase was made basic with 25% NaOH, and extracted with 3x100 mL CH2Cl2. Evaporation of the solvent from these extracts yielded 1.6 g of a clear amber, strong smelling oil which, on distillation at 105-110 °C at 0.2 mm/Hg, yielded 1.0 g of an almost colorless oil. This was dissolved in 20 mL IPA, neutralized with about 10 drops of concentrated HCl, and the spontaneously formed crystals were diluted with 50 mL anhydrous Et2O. After filtration, Et2O washing and air drying, there was obtained 1.1 g white crystals of 3,4-methylenedioxy-N-propargylamphetamine hydrochloride (MDPL) with a mp of 189-190 °C. Anal. (C13H16ClNO2) N.
DOSAGE: greater than 150 mg.
DURATION: unknown.
EXTENSIONS AND COMMENTARY: There is a continuing uncertainty about the name for the three-carbon radical that contains a triple bond. The hydrocarbon is propyne, although it has been referred to as methylacetylene in the older literature. The adjective, going from the triple bond out to the point of attachment, is called propargyl, as in propargyl chloride. When the adjective must be built on the parent hydrocarbon, the double bond is on the outside and one reads away from it, as in 2-propynyl something. However, when the hydrocarbon is essentially the entire structure, then things get named going towards the triple bond, as in 3-chloro-1-propyne. Wait. I'm not done yet° When the actual hydrocarbon name becomes distorted into the derivative, then the triple bond is again at the high end of the numbering scheme. Propynol is 2-propyn-1-ol, which is, of course, the same as 3-hydroxypropyne, or propargyl alcohol. The code MDPL takes the first and last letter of the two of them, both propargyl and propynyl.
#118 MDPR; N-PROPYL-MDA; 3,4-METHYLENEDIOXY-N-PROPYLAMPHETAMINE
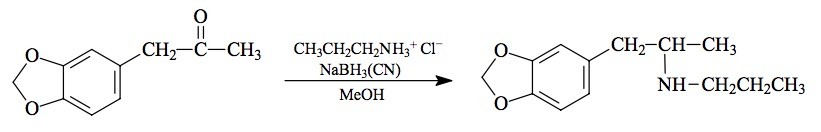

SYNTHESIS: A total of 20 mL concentrated HCl was added beneath the surface of 20 mL propylamine, and when the addition was complete, the mixture was stripped of volatiles under vacuum. The slightly yellow residual oil weighed 20.7 g and set up to crystals on cooling. It was dissolved in 75 mL MeOH, and there was added 4.45 g of 3,4-methylenedioxyphenylacetone (see under MDMA for its preparation) followed by 1.1 g sodium cyanoborohydride. Concentrated HCl in MeOH was added as required to maintain the pH at about 6 as determined with external, dampened universal pH paper. When the generation of base had stopped, the MeOH was allowed to evaporate and the residue was suspended in 1 L water. This was made strongly acidic with an excess of HCl. After washing with CH2Cl2, the aqueous phase was made basic with 25% NaOH, and extracted with 3x100 mL CH2Cl2. Removal of the solvent from the pooled extracts under vacuum yielded 3.3 g of a pale amber oil that was distilled at 85-90 °C at 0.2 mm/Hg. This fraction was water-white and weighed 2.3 g. It was dissolved in 10 mL IPA and neutralized with 25 drops concentrated HCl which produced crystals spontaneously. These were diluted with anhydrous Et2O, removed by filtration, washed with additional Et2O, and air dried. In this way there was obtained 2.3 g of 3,4-methylenedioxy-N-propylamphetamine hydrochloride (MDPR) with a mp of 190-192 °C. Recrystallization from IPA gave a mp of 194-195 °C. The NMR spectrum was completely consistent with the assigned chemical structure. Anal. (C13H20ClNO2) N.
DOSAGE: greater than 200 mg.
DURATION: unknown.
QUALITATIVE COMMENTS: (with 200 mg) There are the slightest hints of physical response, maybe a smidgin of a lightheadedness at the one hour point. Perhaps a slight teeth clench. Certainly there is no central mental effect.
EXTENSIONS AND COMMENTARY: This particular drug, considering that it was without activity, has proven one of the richest veins of pharmacological raw material. Two clues suggested its potential value. A number of reports in the 150 to 200 milligram area suggested that something was taking place in the periphery even without any clear central effects. The term Rbody windowS was used occasionally by experimenters, an outgrowth of the term "window" that was used (at that time, the mid-1970's) to describe the mental effects of MDMA. It was as if the body was opened up and made receptive, instead of the mind. The second clue came from many anecdotal reports that methedrine (a potent central nervous system stimulant) would augment the effects of an LSD dosage which followed it. The putting of a drug on top of an inactive drug is the "primer" concept. It turned out that MDPR was an extraordinary primer to some following psychedelic, especially LSD, even at modest doses. The putting of a drug on top of an active drug, usually during the latter part of its effectiveness is, as previously stated, called "piggy-backing." A third drug-drug interaction has also been studied; the simultaneous administration of two active drugs, to study synergism. There may be an enhancement, or an inhibition, of one with the other. Let's now re-enter the subsection "Qualitative Comments" again, with this primer concept in mind.
QUALITATIVE COMMENTS: (with 160 mg followed at 2 h by 60 5gs LSD) RThe visual phenomena were extraordinary. We were at the beach just south of Mendocino. In anything that had ever been living, there was an endlessly deep microcosm of detail. Endless, and forever more microscopic in intricacy. A sea urchin shell, a bit of driftwood, a scrap of dried seaweed, each was a treasure of jewels. I have never had such wealth of visual eroticism and bliss before. Later, we visited the pygmy forest, but these living fossils were not as magical.
(with 160 mg followed at 2 h by 60 5gs LSD) RWe both felt the first effects at about 30 minutes, and an hour later we found ourselves in a startling folie-a-deux, involved in reliving the origins of man's arrival on earth. We were deep in a tropic environment, defending ourselves against the nasties of nature (insects, threatening things, blistering heat) and determining that man could indeed live here and perhaps survive. A shared eyes-closed fantasy that seemed to be the same script for both of us.
(with 160 mg followed at 2 h by100 5gs LSD) RThis proved to be almost too intoxicating, and a problem arose that had to have a solution. The entire research group was here, and all were following this same regimen. Two hours into the second half of the experiment a telephone call came that reminded me of a promise I had made to perform in a social afternoon with the viola in a string quartet. Why did I answer the phone? My entire experience was, over the course of about 20 minutes, pushed down to a fragile threshold, and I drove about 10 minutes to attend a swank afternoon event and played an early Beethoven and a middle Mozart with an untouched glass of expensive Merlot in front of me. I could always blame the booze. I declined the magnificent food spread, split, and returned to my own party. Safely home, and given 20 more minutes, I was back into a rolling +++ and I now know that the mind has a remarkable ability to control the particular place the psyche is in.
(with 200 mg followed at 2 h by 60 5gs LSD) RThere was a steady climb from the half-hour point to about 2 hours. There was not the slightest trace of anything sinister. There was simply a super tactile person-to-person window. I had an overpowering urge to go out and interact with other people. To see, to talk, to be with others. There are unending fantasies of things erotic. Perhaps being with others should be circumspect. By evening the effects had largely worn off, but this was an incredible day, beautiful and unexpectedly relaxing.
EXTENSIONS AND COMMENTARY: There is need for more commentary. It must be noted that all of the above comments used rather modest dosages of LSD. The notes of this period, some two years of exploring interactions of the MD series of compounds as preludes to true psychedelics, are difficult to distill into a simple pattern. Most of these studies used LSD in the 60-100 microgram range which is fundamentally a modest level. Many trials were made where the challenge of acid plopped right on top of an active residue of another drug was more in keeping with the "piggyback" argument. An illustration of this is a trial in which the primer was MDMA followed at 5 hours (this is at a time of almost no effect) with a larger dose of LSD (250 micrograms). The LSD overwhelmed the residual numbing of the MDMA, and the generated state was overwhelmingly erotic and out of body. There can be no way of analytically organizing such a gemisch of drug-drug interactions with any logic that would allow a definitive interpretation. And LSD is not the only agent that can be used to challenge the Rbody windowS such as that produced by MDPR. 2C-B, 2C-T-2 and 2C-T-7 have all been used with fine success as well.
In general, the use of an MD compound (looking at it as a stimulant and primer) followed by a psychedelic, brings about an exaggeration and enhancement of the latter compound. Much work must be done in this area to make sense of it all.
#119 ME; METAESCALINE; 3,4-DIMETHOXY-5-ETHOXYPHENETHYLAMINE
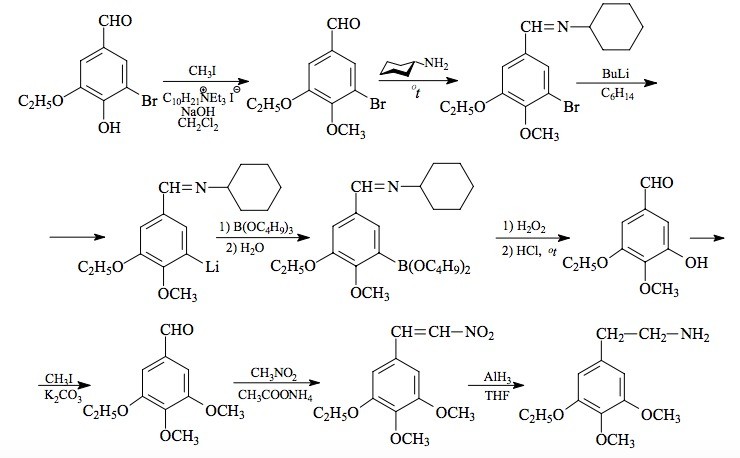

SYNTHESIS: To a vigorously stirred suspension of 18.6 g of 5-bromobourbonal in 100 mL CH2Cl2 there was added 14.2 g methyl iodide, 1.0 g decyltriethylammonium iodide, and 120 mL 5% NaOH. The color was a deep amber, and within 1 min the top phase set up to a solid. This was largely dispersed with the addition of another 50 mL of water. The reaction was allowed to stir for 2 days. The lower phase was washed with H2O, and saved. The upper phase was treated with another 100 mL CH2Cl2, 50 mL of 25% NaOH, another g of decyltriethylammonium iodide, and an additional 50 mL of methyl iodide. The formed solids dispersed by themselves in a few h to produce two relatively clear layers. Stirring was continued for an additional 3 days. The lower phase was separated, washed with H2O, and combined with the earlier extract. The solvent was removed under vacuum to give 20.3 g of an amber oil that was distilled at 120-133 °C at 0.4 mm/Hg to yield 15.6 g of 3-bromo-4-methoxy-5-ethoxybenzaldehyde as a white crystalline solid with a mp of 52-53 °C.
A mixture of 15.6 g 3-bromo-4-methoxy-5-ethoxybenzaldehyde and 10 mL cyclohexylamine was heated with an open flame until it appeared free of H2O. The residue was put under a vacuum (0.5 mm/Hg) and distilled at 148-155 °C yielding 19.2 g 3-bromo-N-cyclohexyl-4-methoxy-5-ethoxybenzylidenimine as an off-white crystalline solid with a melting point 66-68.5 °C. Recrystallization from 100 mL boiling MeOH gave a mp of 67-68.5 °C. The C=N stretch in the infra-red was at 1640 cm-1. Anal. (C16H22BrNO2) C,H.
A solution of 17 g 3-bromo-N-cyclohexyl-4-methoxy-5-ethoxybenzyl-idenimine in 200 mL anhydrous Et2O was placed in an atmosphere of He, stirred magnetically, and cooled with an external dry-ice acetone bath. Then 38 mL of a 1.55 M solution of butyllithium in hexane was added over 2 min, producing a clear yellow solution. There was then added 25 mL of butyl borate at one time, and the stirred solution allowed to return to room temperature. This was followed with 100 mL of saturated aqueous ammonium sulfate. The Et2O layer was separated, washed with additional saturated ammonium sulfate solution, and evaporated under vacuum The residue was dissolved in 200 mL of 50% MeOH and treated with 12 mL of 30% hydrogen peroxide. This reaction was mildly exothermic, and was allowed to stir for 15 min, then added to an aqueous solution of 50 g ammonium sulfate. This was extracted with 2x100 mL CH2Cl2, the pooled extracts washed once with H2O, and the solvent removed under vacuum. The residue was suspended in dilute HCl, and heated on the steam bath for 0.5 h. Stirring was continued until the reaction was again at room temperature and then it was extracted with 2x100 mL CH2Cl2. These extracts were pooled and in turn extracted with 2x100 mL dilute NaOH. The aqueous extracts were reacidified with HCl, and reextracted with 2x100 mL CH2Cl2. After pooling, the solvent was removed under vacuum to yield an oily residue. This was distilled at 118-130 °C at 0.2 mm/Hg to yield 7.5 g of 3-ethoxy-5-hydroxy-4-methoxybenzaldehyde as a distillate that set to white crystals. Recrystallization from cyclohexane gives a product with a mp of 77-78 °C. Anal. (C10H12O4) C,H.
A solution of 7.3 g of 3-ethoxy-5-hydroxy-4-methoxybenzaldehyde in 100 mL acetone was treated with 5 mL methyl iodide and 8.0 g finely powdered anhydrous K2CO3, and held at reflux on a steam bath for 6 h. The solvent was removed under vacuum, and the residue was suspended in H2O. After making this strongly basic, it was extracted with 3x50 mL CH2Cl2, the extracts were pooled, and the solvent removed under vacuum. The residual amber oil was distilled at 110-120 °C at 0.4 mm/Hg to yield 7.3 g of a white oil. This spontaneously set to white crystals of 3,4-dimethoxy-5-ethoxybenzaldehyde which had a mp of 49-49.5 °C. Anal. (C11H14O4) C,H. This same aldehyde can be obtained, but in a less satisfactory yield, by the ethylation of 3,4-dimethoxy-5-hydroxybenzaldehyde described under the preparation of metaproscaline (MP).
A solution of 7.2 g 3,4-dimethoxy-5-ethoxybenzaldehyde in 100 mL nitromethane containing 0.1 g anhydrous ammonium acetate was held at reflux for 50 min. The excess nitromethane was removed under vacuum producing 6.8 g of a red oil which was decanted from some insoluble material. Addition of 10 mL hot MeOH to the decantings, gave a homogeneous solution that spontaneously crystallized on cooling. The yellow crystals were removed by filtration, washed sparingly with MeOH and air dried yielding 3.5 g yellow crystals of 3,4-dimethoxy-5-ethoxy-'-nitrostyrene, with a mp of 89.5-90 °C after recrystallization from MeOH. Anal. (C12H15NO5) C,H.
A solution of 2.0 g LAH in 100 mL anhydrous THF under He was cooled to 0 °C and vigorously stirred. There was added, dropwise, 1.3 mL of 100% H2SO4, followed by the dropwise addition of a solution of 3.1 g 3,4-dimethoxy-5-ethoxy-'-nitrostyrene in 50 mL anhydrous THF, over the course of 10 min. The mixture was stirred at 0 °C for a while, and then brought to a reflux on the steam bath for 30 min. After cooling again, the excess hydride was destroyed with IPA in THF, followed by the addition of 20 mL 10% NaOH which was sufficient to convert the solids to a white and granular form. These were removed by filtration, the filter cake washed with IPA, the mother liquor and filtrates combined, and the solvents removed under vacuum. The residue was added to 150 mL dilute H2SO4, and the cloudy suspension washed with 2x75 mL CH2Cl2 which removed much of the color. The aqueous phase was made basic with 25% NaOH, and extracted with 3x50 mL CH2Cl2. The solvent was removed from these pooled extracts and the residue distilled at 103-116 °C at 0.25 mm/Hg to provide 2.3 g of a colorless viscous liquid. This was dissolved in 10 mL IPA, neutralized with about 25 drops of concentrated HCl, which produced an insoluble white solid. This was diluted with 40 mL anhydrous Et2O added slowly with continuous stirring. The white crystalline 3,4-dimethoxy-5-ethoxyphenethylamine hydrochloride (ME) was isolated by filtration, washed with Et2O, and air dried, and weighed 2.4 g. It had a mp of 202-203 °C which increased by one degree upon recrystallization from boiling IPA. Anal. (C12H20ClNO3) C,H.
DOSAGE: 200 - 350 mg.
DURATION: 8 - 12 h.
QUALITATIVE COMMENTS: (with 200 mg) It tasted pretty strong. However, the taste was soon gone, and an energetic feeling began to take over me. It continued to grow. The feeling was one of great camaraderie, and it was very easy to talk to people. Everyone was talking to everyone else. I found it most pleasant, energetic and at the same time relaxing, with my defenses down. This material did not seem to lead to introspection; however, it might if one took it without other people around. Heightened visual awareness was mild, but the audio awareness was quite heightened. The feeling of being with everyone was intense.
(with 250 mg) Initially I took 200 milligrams of metaescaline, and the experience developed for me very gradually at first, and very pleasantly. After about one half hour I became aware of a wall that seemed to shut me in, not unpleasantly. The wall slowly dissolved, but I was afraid I might get into a negative experience. I felt immediate relief (from this isolation) upon taking the additional 50 mg (at 2:23 into the experiment) as though glad of the decision. I lay down outside on a blanket. There was a marvelous feeling inside, although no imagery. I felt the wall dissolve completely, and I desired to join the group. From this point on the experience was most enjoyable, euphoric. Although not dramatic like some psychedelics, it was most rewarding for me personally. I felt a marvelous bond with everyone present, with clear-headed, excellent thinking, and excellent communication. All in all, a most rewarding and enjoyable experience. Afterwards I felt much strengthened, with good energy and good insight. I have a strong feeling that the group tailored the nature of the experience, and that I and others were most desirous of group interaction. I feel that one could do a lot of other things with it if one turned one's attention to it.
(with 275 mg) Onset of both physical and mental change was slow relative to other psychochemicals. Very gradual internal stirrings were felt at about the hour-and-a-half point. These were mostly feelingful rather than cognitive, and were quite pleasurable. At about the two-and-a-half hour point I grew quite thirsty, and drank a pint of beer. Almost immediately, and quite unexpectedly, I tomsoed to a much higher level and remained there for another three hours until the whole experience waned. [The verb, to tomso, means a sudden rekindling of the drug-induced altered state with a small amount of alcohol. It is explained in the recipe for TOMSO.] During the experience heights, and in fact before it reached its height, talking was easy and unimpeded. The transference feelings so characteristic of MDMA were basically not there. But for purposes of psychotherapy, there were some advantages: fluent associations, undefended positions, and general bonaise.
(with 400 mg) Ingested 300 milligrams at about 1:30 in the afternoon. Very quiet climb. Occasional yawns. Matter-of-fact view of the world. No rosy glow. At the end of the second hour, I seem to be stuck at a ++. Take another 100 milligrams at 3:45 PM. Still tastes awful. Feel a small head-rush fifteen minutes after taking the supplement, and within a half hour I am completely +3. For a while this was a sterner mescaline. Saw the eternal, continual making of choices, all opposites continually in motion with each other. Yin and yang everywhere, giving life to every molecule. The universe itself keeps alive by the action-reaction, the yes-no, the black-white, male-female, plus-minus. All life is a continual making of choices on all levels. Then I closed my eyes, and I found myself floating up to the very top of a temple, where there was radiant light and a sense of homecoming. Making love is a clear stream over and through rocks and canyons Q the earth and sky make love, and the rocks make love to other rocks, and the water is the teasing, fondling, living and moving actions of loving. To realize that, on some level, all existence makes love to all other existence. The Japanese Garden: a structured way of laying out a small glimpse into cosmic love-making, so that it can be read by other human souls. All loving, when direct and free and undemanding, is a touching of the Source. The hardest lesson, of course, is how to love yourself that same way. And it remains both the first lesson of Kindergarten and the Ph.D. final. I was able to drift into sleep at about 4:00 AM.
EXTENSIONS AND COMMENTARY: The reorientation of the single ethyl group of escaline (E) to the meta-position produces metaescaline (ME). In cats, in studies of over 50 years ago, the two compounds produced similar effects at similar dosages. In man, ME also appears to be similar to mescaline in potency. However, a subtle difference is apparent between ME and Peyote, the natural source of mescaline. With Peyote itself, the initial taste of the crude cactus is more than just foul; it might better be described as unbelievably foul. But in the middle of a Peyote experience, the taste of the cactus is truly friendly. When ME was retasted in the middle of an experience, the taste was still foul.
There are other distinctions from mescaline. Unlike mescaline or Peyote, there is rarely any body discomfort during the early phase of intoxication, no nausea and only an occasional comment suggesting hyperreflexia. And, also unlike mescaline, most subjective reports on ME claim that music produces little imagery, and the exaggeration of color perception is more reserved. Appetite is normal, the tastes and textures of food are unusually rewarding. No subject has ever expressed a reluctance to repeat the experience. Sleep is easy, refreshing, and the following day seems free from residue.